
Some equipment or products are so sensitive that the slightest invisible contaminant can harm them. In places such as data centers or pharmaceutical facilities, these contaminants should be well monitored and eliminated, otherwise, they could disrupt operations or endanger consumers. Ensuring that cleanrooms are free from contaminants is one-way companies can earn the trust of clients and get ahead of the competition.
What Is a Cleanroom?
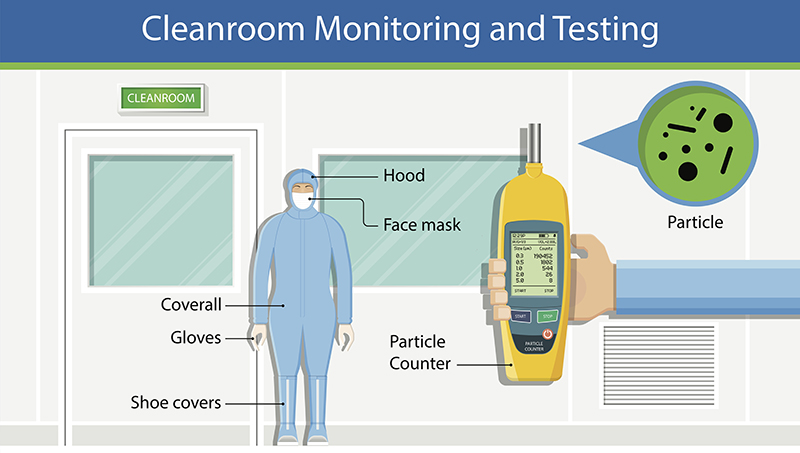
Cleanrooms, or clean zones, are critical locations usually found in industries such as pharmaceuticals, healthcare, data centers, research facilities, and microelectronics. They are environments that are guarded against atmosphere contaminants such as dust, carbon, airborne microbes, aerosol particles, microorganisms, and chemical vapors. These contaminants are potentially dangerous to pharmaceuticals and bio-pharmaceutical products, biomedical, electronic devices, or scientific research and other sensitive manufactured products.
How Does Cleanroom Monitoring Work?
Since contaminants present dangers to different parts of the facilities, monitoring cleanrooms is a vital process. Monitoring, controlling, and reducing air contamination will help ensure the quality and integrity of the products. Environmental conditions such as humidity, temperature, differential pressure, and airflow can also be monitored in cleanrooms, as well as air quality.
In planning a cleanroom monitoring, here are the steps:
Understand and Implement Industry Guidelines
Of course, cleanrooms should not only be clean, above all, they must also be compliant. Available standards and regulations can provide more detailed information about cleanroom requirements. Here are some of typical standards and regulations, across different areas:
ISO 14644-2: 2015. This regulation specifies minimum requirements for monitoring plans for cleanroom or clean zone performance. This is related to air cleanliness by observing particle concentration, based upon parameters that measure airborne particle concentrations. This is the only ISO standard regarding monitoring of air cleanliness by particle concentration. The latest document, the 2015 version, is now offering a risk-based approach, with a section about “creating, implementing and maintaining a monitoring plan.”

EU GMP Annex 1. These are rules that govern medicinal products within the European Union. In the EU GMP Annex 1, a section called “Cleanroom and clean air device monitoring” offers information and ways to achieve the requirements.
FDA Guidance for Industry Sterile Drug Products Produced by Aseptic Processing. This US agency offers extensive information on environmental and particle monitoring. This guidance is intended to help manufacturers meet the requirements in the Agency’s current good manufacturing practice (CGMP) regulations (2l CFR parts 210 and 211) when manufacturing sterile drugs and biological products using aseptic processing.
World Health Organization (WHO) Environmental Monitoring of Clean Rooms in Vaccine Manufacturing Facilities. These are vaccine-specific cleanroom monitoring guidelines. These guidelines contain vital information regarding routine monitoring and sampling plan.
PDA Technical Report (TR) No.13R Fundamentals of an Environmental Monitoring Program. This document serves as a resource for controlled environmental test methods, although some nonviable particulate information is included, the report’s primary focus is microbiological control for sterile product manufacturing. It is expanded from PDA’s 2001 revision of Technical Report No. 13, to reflect substantial changes to regulatory guidelines, international standards, and scientific advances in environmental monitoring procedures and equipment. TR-13R is one of the best documents to achieve a proper monitoring plan because it summarizes requirements that must be followed, and offers sample plans.
Assess Risks Based on Probability and Severity
For correct monitoring solutions, management should assess the risks associated with cleanrooms. Risk is defined as the combined probability of the occurrence of harm, and the severity of that potential harm. Risk assessment is the identification of hazards and the analysis of risk associated with those hazards.
One of the best risk management guidelines comes from the ICH guidelines on Q9 Quality Risk Management. This document can help prepare an execution roadmap. According to the document, the company should ask the following: What might go wrong? What is the likelihood (probability) it will go wrong? What are the consequences or severity if events to go wrong?
Some guidelines, such as ISO 14644-2, require an appropriate risk assessment tool as the first step of the risk assessment. Some of them are:
- HACCP – Hazard Analysis and Critical Control Points
- FMEA – Failure Mode Effects Analysis
- PHA – Preliminary Hazard Analysis
- FTA – Fault Tree Analysis
- HAZOP – Hazard and Operability Analysis
At Lighthouse Worldwide Solutions, for instance, they generally use two tools. For non-viable monitoring or monitoring of particles that don’t contain microorganisms, they use the Hazard Analysis and Critical Control Points (HACCP), and to rank the urgency of the monitoring points, they use Failure Mode and Effects Analysis (FMEA).
When using the FMEA as the risk assessment tool, it’s better to ask and score the probability and severity first:
Probability – These questions should be asked: What is the probability of having particles at this location that will harm the product, which will end up affecting patient health? For instance, with regards to a turntable after a dehydrogenation tunnel, containers are exposed to ambient air, and having particles in supposed sterile containers may lead to HVAC failure. In this case, the probability is very high.
Severity – In scoring the severity, the same procedure applies. The questions that can be asked: If they have particles within the container, how did it end up there? What will be the result? Is it fine or will it be damaging? In this case, the severity is high. High probability with high severity results in a Level-1 risk level. In reducing risks, this Level-1 should be the main focus.
Define Sample Locations
This is the next step after defining risks. After knowing the situation of the facility in terms of probability and severity, then it’s time to know the sample locations. Before doing that. management has to reduce associated risks. Start identifying the highest risk locations, then move towards lower-risk zones. Below are considered high-risk locations in sterile pharmaceutical manufacturing facilities:

- After tunnel exit/turntable
- Point of fill
- Stoppering
- Product transfer hatches/window for semi- stoppered lyophilized products
- Transfer CART’s from/to Lyophiliser
- Lyophiliser loading
- Capping zone to reduce risk due to poorly closed stoppers after lyophilization
- Room Grade B, including any location based on the product, personnel flow, and room design
- Autoclave and sterilizers exit to Grade B areas
- Sterile/aseptic manipulation zones, such as aseptic connection and under laminar airflow and Grade
If opting for a particle counter, which is a good choice, one of the best practices is placing them close, or right under the sample point. This can help count the particles because they will not be lost due to travel distance.
Here are some things to consider:
It’s important to conduct an engineering study. Some locations may require additional investigation, especially if more statistical data needs to be collected between different sample points for the same risk locations.
It’s important to consider different solutions. For particle monitoring, several options are available. For example, built-in pump particle counters or external vacuum systems, devices using analog or digital sensors, with and without display, are available. The most important feature that management should be looking for is self-diagnostics. This means that the particle counter should not only collect data one-way, but also be able to communicate flow status, laser health, and internal parameters such as background voltage and calibration of due date reminders.
Conclusion
Planning a cleanroom monitoring strategy is important, as they ensure the stability of monitoring solutions. These plans will lead to better understandings of air quality and room conditions. Most importantly, this process ensures that the monitoring systems and the cleanrooms are compliant with industrial regulations.
AKCP provides monitoring solutions that can be used in cleanrooms for the pharmaceutical, food processing and electronics manufacturing industries.



